
My daily 6am workout/data analysis sessions
I’ve been running a lot of experiments these past few weeks to try to make up for a conference I’ll be attending (the 2019 Research Society on Alcoholism meeting.) so I’ve been generating a lot of data and wanted to share what a typical morning looks like. If you’ve been keeping up with my posts recently, I’ve been doing A LOT of cloning. To verify I have the correct DNA I think I made, I need to analyze the DNA sequence (see this blog post on how this data is generated.) I wanted to upload a snippet of the analysis process. I wake up at 5-6am everyday and spend 2 hours working out every day, but since I put a laptop stand on my bike/treadmill, I’m also able to analyze data and plan experiments (I don’t like going into lab not knowing what to do.)
6-18-19 analysis:
T7 sequencing of pLR-NR2B 6-14-19 shows that I have a 5’UTR, but no start sequence, since the primer for the backbone did not include a gene-specific sequence. T3 sequencing show that I do in fact have the very c-terminus of NR2B, but that after about 486 BP before the stop codon, the 5’UTR binds to the NR2B gene. T7 sequencing is a little short of perfect since its so close to the 5’UTR gene, but it looks like the product from this sequencing result might be usable; it has the minimum things I need: a 5’UTR and 20-25BP homology to the 3’ end of the original, un 3’UTR’d PolyA’d end. Can technically combine this with the pBS-MST rev primer and the open pLR2B-F primer (20bp), since sequencing confirms that both sequences are perfectly there there. For pLR2B-6-15-19 #3 T7 sequencing says its actually pJS2B. Did not DpnI digest so it makes sense. For pLR2B-6-15-19 #2 it was too low in conc. to work. For pLR2B-6-16-19 #1, somehow this one annealed in on itself to just after orf3, which is the ampicillin resistance region.
T1pLR2B#3 technically has ATGAAGCCCAGCGCAGAGTGCTGTTCCCCCAA, which is 3 amino acids after the 5’ UTR-pLR2B forward primer. Perhaps the 5’ 2B primer is sticky/loose? Or maybe the reverse primer for the backbone is strange. Similar problem to the pLR-NR2B 6-14-19; if I did T3 sequencing (I didn’t) and it was matched to NR2B, same strategy could be used: pBS-MST Rev + pLR2B Open FW PCR (can’t use the pLR backbone primers, too much homology increases Tm too much). There is a reasonable chance T1-pLR2B#2 worked for pLR2B (5’UTR only, no 3’UTR/PolyA yet). I was thinking that statistically, SOMETHING should’ve worked. Must double check the DNA gel results and the concentrations obtained on the nanodrop, because I remember that yesterday, it seemed like nothing at all worked. That said, whatever is there is present at a concentration reasonable for sequencing, right? Plus, IF T1-pLR2B #2worked, THEN pLR-open FW + pLR-2B Rev should give me a 7.X kB product. Easy enough to run on a gel and verify. For T1-pLR2B #1, same issues as T1-pLR2B #3, I see a sequence (CTCAACTTTGGATGAAGCCCAGCGCAGAGTGCTGTTCCCCCAAGTTCTGGTTGGTGTT) that says the 5’UTR+ several amino acids coding for my gene, but then it abruptly starts going to the c-terminal NR2B sequence. Same conclusion and possible strategy. Alternative strategy: lower SLIC digest time. Less ssDNA means less likelihood of off-target products.
Conclusions: Throw away the innoculants/minipreps from 6-15-19 and 6-16-19. Despite not getting complete 5’UTR+3’UTR/PolyA products, I technically do have the starting materials to proceed to two possible SLIC experiments:
1) PCR amplify pLR-NR2B 6-14-19 with pBS-MST Rev + pLR2B Open to produce a pLR backbone. The backbone could then be combined with 5’UTR-pLR-2B-3’UTR PCR product from before, + PolyA and 3’UTR dsDNA. I prefer pLR-NR2B 6-14-19 because I have T7 and T3 sequencing that says I have what I think I do. Double check the DNA gels I ran and the nanodrop concentrations. If the yield is bad or if there are other concerns (off target products affecting nanodrop results) then proceed with T1pLR2B#3 (or #1, check DNA gel and nanodrop) PCR instead, just send sample for sequencing (T3). The size of the band on the gel can be wrong, I know I have the bare minimum for PCR to work (on paper).
2) Carefully analyze the gel of T1-pLR2B#2 and the nanodrop results. Consider running it on a DNA gel and gel extracting the product if its present in good quantity. Can inoculate this into bacteria. Then, perhaps PCR amplify T1-pLR2B#2 with pLR-open FW + pLR-2B Rev. DpnI digest (overnight) then SLIC with 3’UTR and PolyA. The size of the band on the gel is important here. I don’t know what I have past ~966 BP. That said, why would this fail at a different part of the experiment when it succeeds where others could not (5’UTR+NR2B). Carefully analyze the gel of T1-pLR2B#2 and the nanodrop results. Consider sending it off for sequencing, using T3 and a few NR2B primers.
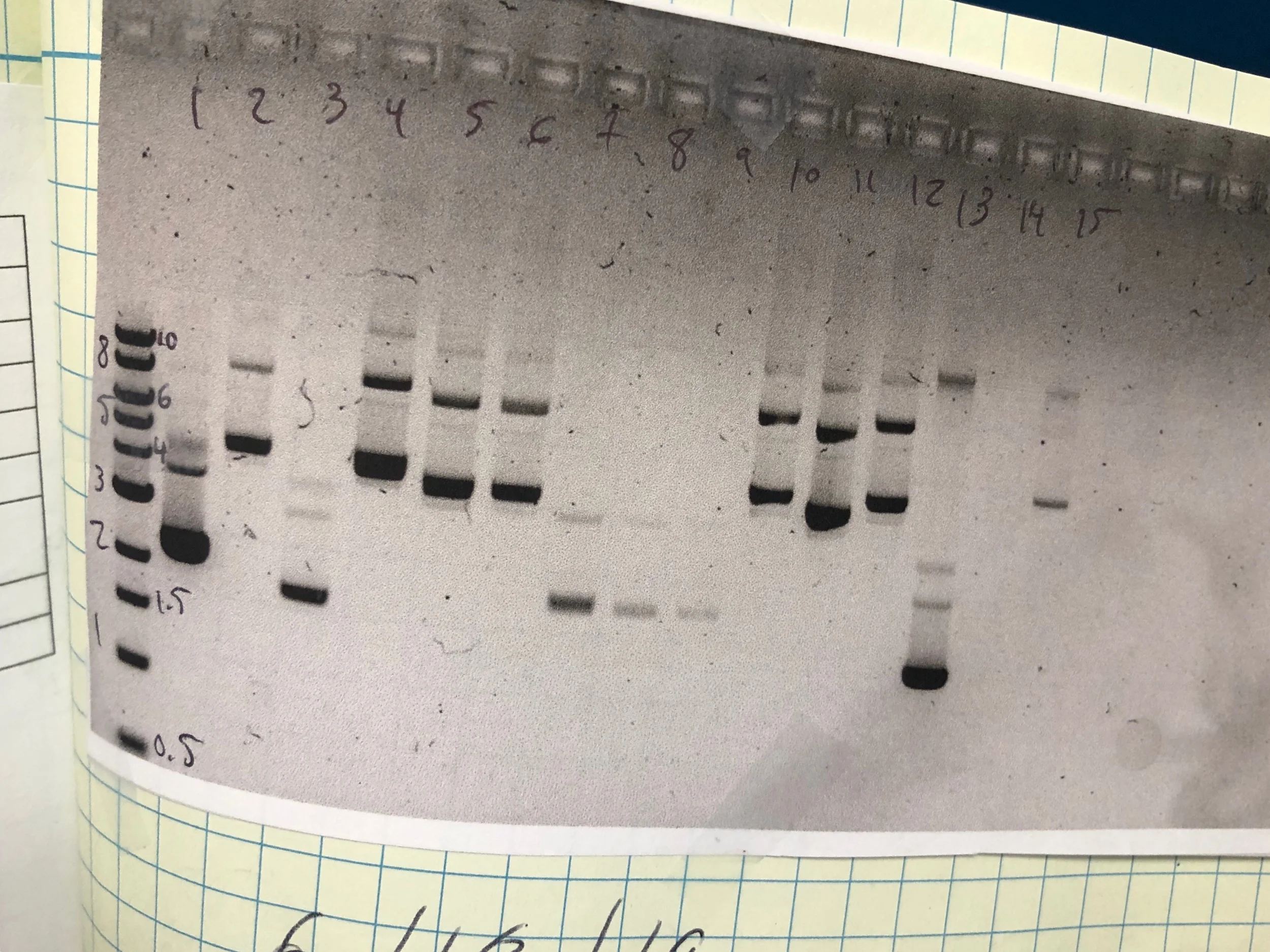

6-20-19 update (still on the treadmill right now):
I’m completed the PCRs described above and will be performing SLIC today. I’m still skeptical about the T1pLR2B#2; it consistently runs at ~5-6kB. Worse, supercoiled (plasmid) DNA should run HIGHER than the anticipated size. If you mess with the contrast on the gel, you can see a band just below 8kB. In the gel above, you can see a larger band above 8kB. We could run all the DNA from the miniprep on a gel, then gel extract the above products and transform into bacteria. Technically we should be getting only one band, so in this regard, this is a good strategy to get more product. That said, why is T1pLR2B#2 coming back as having most of the NR2B insert if the size runs wrong? TECHNICALLY the sequencing primers could bind to the correct product IF its present in the miniprep, and the sequencing data looks clean. However, we see from the nanodrop that T1pLR2B#2 had a low DNA yield; from the gel, it doesn’t look like the products >6kB could be present in large enough quantities to give a semi-false positive. If its good enough to sequence correctly, is it good enough to produce the right PCR product for a SLIC experiment?
Caveat: I can’t sequence the full length plasmid because its such a low yield.

